Abstract
Cerebral vascular injury is a common phenomenon after traumatic brain injury (TBI), with complications including vascular inflammation, decreased cerebral blood flow, and increased vessel tortuosity. Promoting cerebrovascular health may therefore be a useful therapeutic intervention after TBI. ApoA-I, the major protein constituent of circulating high-density lipoproteins (HDL) are an attractive target due to its vasoprotective and anti-inflammatory roles in periphery vessels, but little is known on whether these benefits extend to the brain. To address this gap in knowledge, this study was designed to test the novel hypothesis that ApoA-I deficiency would worsen acute vascular and inflammatory outcomes in mice after Closed-Head Impact Model of Engineered Rotational Acceleration (CHIMERA) TBI. ApoA-I Knockout (ApoA-I KO) and WT mice underwent single moderate-severe TBI. Histopathological outcomes at 6hr and 2 days (2D) post-injury were assessed. ApoA-I KO mice exhibited greater Intercellular Adhesion Molecule 1 (ICAM-1), a marker for vascular permeability, in the cortex at 2D post-TBI compared to WT controls, and a subtle increase in brain pro-inflammatory cytokine levels. These results suggest the role of ApoA-I in protecting against TBI induced inflammation.
Introduction
Cerebral vascular injury is a nearly universal event in traumatic brain injury (TBI), where a functional deficit of the neurovascular unit (NVU) is sustained either directly due to the initial physical injury or occurs over time as part of the following secondary inflammation cascade. (Kenney et al., 2016). The NVU is comprised of endothelial cells, basal lamina covered by smooth muscle cells (replaced by pericytes in capillaries), astrocytes, neurons, and the extracellular matrix, which interact as a major conduit in maintaining central nervous system (CNS) homeostasis, among other functions (Salehi, Zhang, & Obenaus, 2017; Kenney et al., 2016; Muoio, Persson, and Sendeski, 2014).
In particular, damage to the cerebral vasculature often results in neuroinflammation (Schimmel, Acosta, & Lozano, 2017; Elder et al., 2015). Damage to the vasculature can alter the extracellular matrix, promoting the infiltration of peripheral leukocytes into the CNS and activating resident glial cells, including astrocytes and microglia (George & Geller, 2018). Chronic activation of glial cells in the brain contribute to elevated pro-inflammatory cytokine levels, including IL-6 and TNF-α (Schimmel, Acosta, & Lozano, 2017; Elder et al., 2015), and secondary cell death in the cascade of TBI sequelae (Schimmel, Acosta, & Lozano, 2017). These elevated pro-inflammatory cytokines in turn lead to an increased expression of adhesion factors, such as ICAM-1 in astrocytes, microglia and endothelial cells alike, which enables increased leukocyte binding to the endothelium. The increase in leukocyte binding lead back to increased efflux of immune cells from the blood vessels to the brain parenchyma, completing the vicious cycle of transendothelial leukocyte extravasation and pro-longed neuroinflammation (Lutton et al., 2017; Balabanov et al., 2001; McKeating & Andrews, 1998; Shrikant et al., 1995).
The strong association of vascular injury with TBI has prompted an exploration into the linkage between general vascular health and injury outcome. High-density lipoprotein (HDL) has well -defined vasoprotective effects in peripheral blood vessels outside of the brain , with low plasma levels of HDL firmly established as a predictor for cardiovascular disease (Besler, Lüscher, & Landmesser, 2012; Mahdy Ali, Wonnerth, Huber, & Wojta, 2012). ApoA-I, the major protein component of circulating HDL in plasma, has been shown to reduce inflammation through the inhibition of macrophage chemotaxis and reduced monocyte recruitment from the circulation to the tissue (Iqbal et al., 2016; Yin et al., 2011). It does this by the suppression of adhesion molecules expressed on endothelial cells (Calabresi et al., 1997). In addition, it promotes endothelial nitric oxide (NO) synthase activity, thereby reducing inflammation via downregulation of the pro-inflammatory NFκB signalling pathway (Boyce, Button, Soo, & Wellington, 2017).
ApoA-I is produced by hepatocytes and enterocytes in the liver and small intestines respectively. Although ApoA-I mRNA is not expressed in the brain (Elliott, Weickert, & Garner, 2010), its protein is detected in the brain and in cerebrospinal fluid (CSF). While it is unclear whether the protective effects of peripheral ApoA-I is shared in the brain, recent evidence supports the role of HDL in reducing neuroinflammation in mice models. This includes the finding of increased vascular ICAM-1 expression as well as increased vessel-associated astrogliosis among ApoA-I KOmice when compared to mice hemizygous for ApoA-I (Button et al., 2019).
While these results suggest that peripheral ApoA-I may also exhibit vasoprotective properties in the CNS, whether ApoA-I can offer protection from TBI-induced brain changes has not been tested. To investigate the protective role of ApoA-I, we utilized a previously published CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration) model of TBI, developed by our group, (Cheng et al., 2019; Vonder Haar et al., 2019; Namjoshi et al., 2014, Bashir et al.,2020) to induce a closed-head, surgery-free and clinically relevant TBI to ApoA-I Knockout (ApoA-I KO) and wild type (WT) mice. In this study, we tested the novel hypothesis that ApoA-I KO would worsen acute vascular and inflammatory outcomes in mice after CHIMERA TBI as indicated by increased pro-inflammatory brain cytokine levels and vascular adhesion factors. Overall, this study helps shed light on the function of ApoA-I in the brain and by extension, uncovering HDL as a potential therapeutic in improving post TBI inflammatory outcomes.
Methods
Animals and CHIMERA procedure
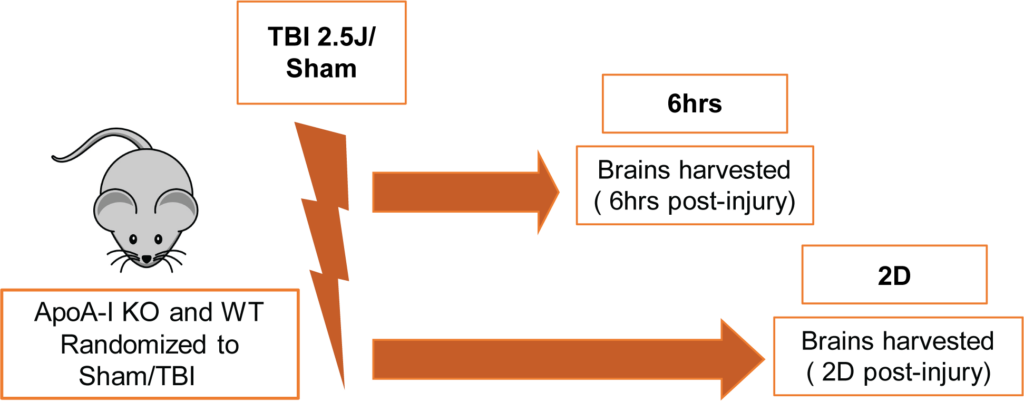
All procedures involving animals were approved by the Canadian Council of Animal Care and the University of British Columbia Animal Care Committee. Male and female ApoA-I KO [B6.129P2-Apoa1tm1Unc/J] and WT [C57BL/6J] mice (total N=64) were purchased from The Jackson Laboratory. Animals were 3.5-5 months of age at the time of impact and were randomized into sham or TBI groups of 6hr or 2D endpoints. The TBI group received an impact energy of 2.5 J whereas the sham group received anesthesia and restraint but no impact. A schematic of the experimental timeline is shown in Figure 1. The CHIMERA model was used to induce TBI as described previously (Bashir et al.,2020). Two ApoA-I KO mice (10% of injury group) died right after CHIMERA procedures due to apnea and no WT animals expired prematurely. The groupings for all animals kept for final analysis were;
WT-6hr (Sham:N=6 (3 Male, 3 Female), TBI:N=8 (5 Male, 3 Female)); WT-2D (Sham:N=6 (3 Male, 3 Female), TBI:N=7 (4 Male, 3 Female)); ApoA-I KO-6hr (Sham:N=7 (3 Male, 4 Female), TBI:N=9 (5 Male, 4 Female)); ApoA-I KO-2D (Sham:N=8 (4 Male, 4 Female) , TBI:N=11(5 Male, 6 Female)) respectively.
Tissue Collection
Mice were fasted for 4 hours prior to anesthetization by intraperitoneal injection of 20 mg/kg xylazine (Bayer, Whippany, NJ, USA) and 150 mg/kg ketamine (Zoetis, Florham Park, NJ, USA). Once a surgical plane of anesthesia was reached, mice were perfused for 7 min with ice-cold phosphate-buffered saline (PBS) containing 2500 U/L heparin at 6 mL/min. Brains were excised and bisected in the sagittal plane. Half of the brain used for biochemical analysis was snap-frozen on dry ice and stored at −80°C until use. The remaining half-brain was fixed in 4% paraformaldehyde (PFA) for 2 days at 4°C followed by cryoprotection in 30% sucrose and 0.1% sodium azide at 4°C, after which 20-μm-thick coronal sections were cut using a cryotome (Leica Microsystems, Buffalo Grove, IL, USA).
Cytokine ELISA
Half-brain lysates were homogenized in 8-volumes of ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer containing protease and phosphatase inhibitor cocktails (Roche, Branford, CT), and centrifuged at 9000 rpm for 10 min at 4 °C. The supernatant was extracted and frozen at −80 °C until analysis. Inflammatory cytokines interleukin 6 (IL-6), interleukin-12 (1L-12), keratinocyte chemoattractant/ human growth-regulated oncogene (KC/GRO), tumour necrosis factor α (TNF-α) levels were measured in RIPA lysates using MesoScale Discovery ELISA kit (K15048G, MesoScale Discovery, 1:2) according to the manufacturer’s instructions. Data points were interpolated from a standard curve and normalized to total soluble protein concentration measured by bicinchoninic acid (BCA) assay (Thermofisher, 23225) per the manufacturer’s instructions.
Histology
Immunofluorescence microscopy was used to visualize intercellular adhesion molecules (ICAM-1) and their colocalization with endothelial cells (CD31). One section from the dorsal hippocampus and one section from the ventral hippocampus were selected from each cyrosectioned half-brain, which were mounted on Superfrost Plus slides. Antigen retrieval was performed with citrate buffer followed by washing with PBS, permeabilizing tissue with Triton, and blocking with 5% donkey serum, 1% BSA and 0.3% Triton X-100 in PBS for 60 minutes. Primary antibodies, ICAM-1 (R&D, AF796, 1:50) and CD31 (Abcam,ab28364, 1:200) were incubated overnight at 4°C, followed by 3x phosphate buffered saline (PBS) washing steps. Next, incubation with secondary antibodies donkey anti-goat Alexa 594 (Invitrogen, A-11058, 1:400) and donkey anti-rabbit Alexa 647 (Invitrogen, A-31573, 1:400), for ICAM-1 and CD31, respectively at room temperature for 2 hours. The samples were then washed 3x in PBS, cover-slipped and mounted onto slides in Prolong Gold Antifade with DAPI (Invitrogen, P36931). The slides were stored at 4°C until imaging with an Axio Scan.Z1 (Zeiss) slide scanner.
Image analysis
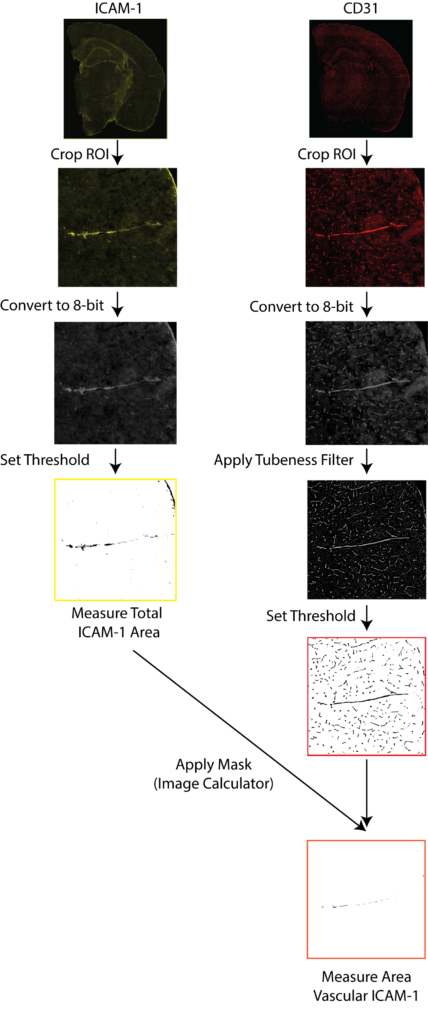
Entire coronal sections were imaged with Zeiss Axio Scan.Z1 (Carl Zeiss Microscopy, Thornwood, NY, USA) at 20× magnification, using fluorescent (ICAM/CD31) imaging. Downstream image analysis was performed with ImageJ (NIH)
A schematic of ICAM-1/CD31 quantification is illustrated in Figure 4. Exported images for each channel were converted to 8-bit black and white images. Cortical regions in the anterior hippocampus and the entorhinal cortex from the posterior hippocampus were drawn manually and saved as regions of interest (ROI) for each sample, using the Allen Mouse Brain Atlas as a guide. ICAM-1 signal was quantified by thresholding and reporting the percentage area containing signal relative to the total cortical area after filtering a background noise <15 pixels. Stained vessels from the CD31 channel were selected with the ImageJ macro Tubeness. The outputs of Tubeness were thresholded and masked, after filtering of background noise <100 pixels, CD31 percentage area was normalised to selected ROI areas. Vascular ICAM-1 was quantified as the ICAM-1 positive area associated with CD31 in each region where masks of the segmented CD31 image for a section were applied to the segmented ICAM-1 image of the same section, using the ImageJ macro ImageCalculator. The ICAM-1 positive area within the CD31 mask was measured then normalized to the total CD31-positive area within the ROI.
Statistics
All animal groupings were blinded during analysis by using surrogate identifying codes. Statistical analyses were performed using IBM SPSS Statistics version 23 software (IBM, Armonk, NY, USA), and graphs were plotted using Prism version 6.07 software (GraphPad Software, La Jolla, CA, USA). For the analysis of histological data, 3-way ANOVA was used considering ApoA-I genotype as one factor, injury status as another factor and timepoint of injury as the third factor, followed by Sidak’s multiple comparisons test if significant factor or interaction effects were observed. For inflammatory cytokine assays, Kruskal-Wallis non-parametric test was used to detect overall effect, followed by Bonferroni corrected Mann-Whitney test to detect meaningful between-group differences.
Results
ApoA-I deficiency enhances the acute inflammatory response after CHIMERA TBI
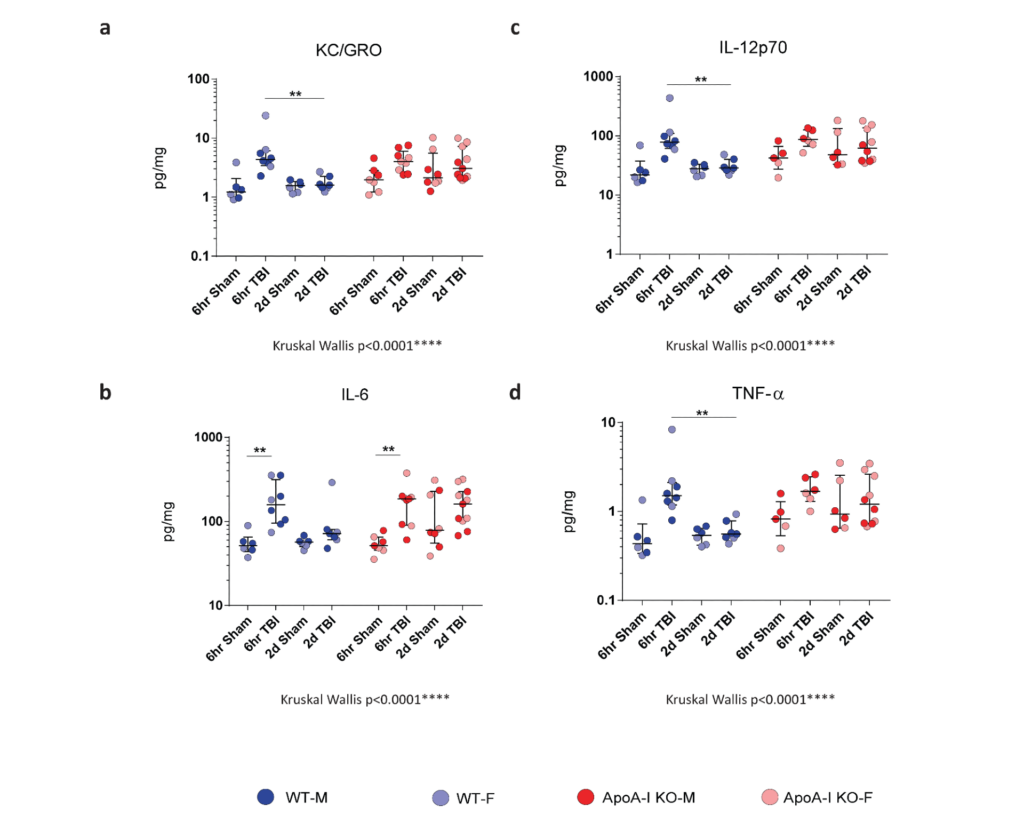
To assess differences in inflammatory status in brain tissue after TBI, we examined cytokine levels in RIPA brain lysates via Mesoscale cytokine ELISA. IL-6 and TNF-α are recognized markers of acute inflammation following TBI, and are produced by activated immune cells. KC/GRO is involved in neutrophil activation and subsequent activation of cells of the innate immune response. IL-12p70 is produced by dendritic cells, macrophages and neutrophils, and is involved in the activation of naïve T cells to activated T cells. We observed a significant overall effect in all four cytokines by Kruskal-Wallis analysis. Further, Bonferroni adjusted Mann–Whitney post-hoc analysis revealed a significant increase in IL-6 levels for both WT (p=0.002) and ApoA-I KO mice (p=0.008) at 6hr post TBI (Figure. 2b). A trend for a significant injury driven increase was observed for WT TBI animals at 6hrs for KC/GRO, IL-12p70 and TNF- α (p=0.056), while no significant increase in measured cytokine levels was observed for ApoA-I KO animals at 6hrs (Figure. 2 a,c,d).
There was a significant decrease in cytokine levels between the WT 6hr TBI group and WT 2D TBI group (p=0.007) for KC/GRO, IL-12p70 and TNF- α (Figure 2a,c,d). No difference of pro-inflammatory cytokine levels between the WT sham and WT 2D TBI group could be detected (Figure 2a,c,d), suggesting that the inflammatory response was resolved by 2D in WT TBI mice. Intriguingly, the same pattern was not observed in ApoA-I KO mice as cytokine levels remained elevated between the 6hr TBI and 2D TBI group (p=0.456, p=1.000,p=0.368,p=0.368 for KC/GRO, IL-6, IL-12p70, and TNF-α respectively (Figure. 2a-d). Of note, data points for pro-inflammatory cytokine levels in ApoA-I KO animals were higher and spread over a wider range compared to WT animals regardless of injury status (Figure. 2a-d), suggesting a higher baseline cytokine level relative to WT. In addition, while no significant increase was observed in comparing the ApoA-I KO 2D TBI against the WT 2D TBI groups, there was a trend towards significance for KC/GRO (p=0.073), IL-12p70 (p=0.085),TNF-α (p=0.085) respectively. Figure(a,c,d).
Taken together, while is it clear that WT animals experienced an initial heightened inflammation at 6hr post-injury, this response was mostly attenuated at 2D. This temporal injury-recovery pattern, however, does not appear to be similar in ApoA-I KO animals.
ApoA-I deficiency increases injury driven total ICAM-1 positive area at the cortex at 2D post-TBI
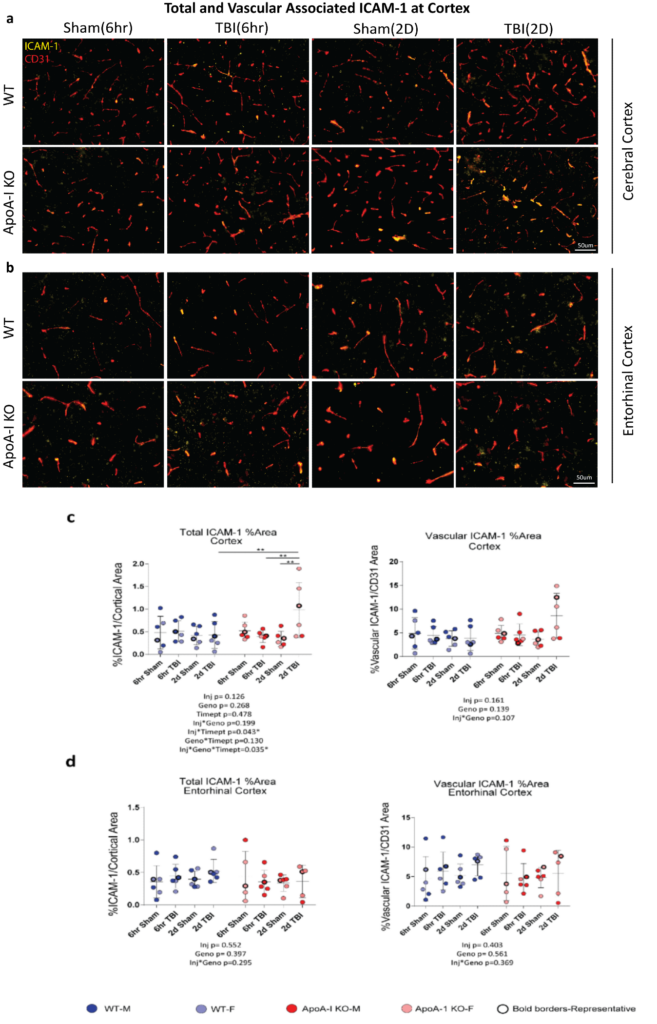
To better analyze specific brain areas affected by TBI, we examined the presence of inflammatory markers through immunofluorescence. ICAM-1 is an adhesion molecule produced by endothelial cells, astrocytes and microglia, and is a prominent marker for vascular inflammation as it promotes leukocyte adhesion to endothelial cells. We quantified the ICAM-1 signal in the entire cerebral cortex, spanning the retrosplenial area to the auditory areas where blood vessels are abundant, and observed a significant injury x genotype x timepoint effect (p=0.035) in total ICAM-1 area by 3-way ANOVA (Figure. 3 a,c). Sidak adjusted post-hoc analysis revealed increased percentage stained total ICAM-1 between the 2D Sham ApoA-I KO and 2D TBI ApoA-I KO animals (p=0.001), from a mean of 0.343 to a mean of 0.981, while levels in WT animals remained consistent regardless of injury status or timepoint (p=0.863 for 6hr Sham WT vs 6hr TBI WT, p=0.935 for 2D Sham WT vs 2D TBI WT) (Figure. 3c). In addition, although main group differences were not significant for vascular associated ICAM-1 (Figure. 3c), there was a trend of increased vessel-associated ICAM-1 in 2D TBI ApoA-I KO animals compared to other groups. This finding demonstrated that, compared to WT animals, ApoA-I KO animals with TBI had a stronger induction of cortical ICAM-1, suggestive of a more pronounced inflammatory status. However, specific examination of the entorhinal cortex revealed no significant injury or genotype differences for both total ICAM-1 and vascular associated ICAM-1 in this region (Figure. 3d). Together, these data suggest the potential for ApoA-I deficiency to exacerbate TBI-induced inflammation at the cerebral cortex at 2D, although more studies are needed to validate this preliminary observation.
Discussion
This is an exploratory study of the potential role of ApoA-I in acute responses to TBI, with a pre-determined focus on cerebrovascular outcomes as ApoA-I is implicated in vascular contributions to Alzheimer’s Disease (AD).
Our results provide proof-of-concept data that ApoA-I deficiency may alter the acute inflammatory response after moderate CHIMERA TBI. We observed a significant increase of cortical ICAM-1 total expression at 2D post-TBI for ApoA-I KO animals only (Figure 3), and an overall increase in cytokine levels independent of injury or timepoint status for ApoA-I KO animals (Figure 2). An increase in total ICAM-1 immunofluorescence was aligned with elevated cytokine levels observed in ApoA-I KO animals as cytokines can lead to endothelial activation, resulting in an increase in expression of adhesion factors (Lutton et al., 2017; Balabanov et al., 2001; McKeating & Andrews, 1998). However, histological data suggest a highly regional-specific difference, as the increase in ICAM-1 expression was only observed in the cerebral cortex at the very acute timepoints studied, though this may be due to how the CHIMERA impact is made beneath the cerebral cortex. Since no baseline differences in ICAM-1 staining between ApoA-I KO and WT animals were observed in this study (Figure 3), ApoA-I deficiency may have a role in the inflammatory response during the recovery process after injury. Supporting this conclusion is the observation that ApoA-I KO animals did not follow the typical injury-recovery temporal response observed in WT animals (Figure 2). Notably, data points for cytokine levels in ApoA-I KO animals were spread over a wider range compared to WT animals regardless of injury status (Figure 2). This may imply that the higher variation in the ApoA-I KO group hindered detection of the cellular response to injury and subsequent recovery. Importantly, these results will need to be confirmed or refuted in future studies with additional animal numbers to provide more statistical power.
This study agrees with previous results published using moderate CHIMERA, in that cytokine elevation was observed at 6hr only for WT animals, and that histological evidence indicative of inflammation was most robust at 2D post TBI (Bashir et al.,2020).
The most surprising finding was that ApoA-I deficiency appeared to affect grey matter more robustly than the vasculature, as we observed an increase in total cortical ICAM-1 rather than CD31(vascular) associated ICAM-1 following TBI. This result is contradictory to what our group has observed in APP/PS1 mice, a common Alzheimer’s Disease (AD) animal model, where APP/PS1 mice deficient in ApoA-I had increased vascular ICAM-1 adhesion as well as increased vessel-associated astrogliosis when compared to APP/PS1 WT animals (Button et al., 2019). This may be due to the fact that APP/PS1 mice express and accumulate Aâ in the brain parenchyma and in cerebral vessels which triggers the production of increased ICAM-1 in the endothelial cells (Verbeek et al., 1994). Indeed, ICAM-1 is expressed in astrocytes, microglia and endothelial cells alike (Dietrich, 2002). Our results may imply that ApoA-I deficiency enhances ICAM-1 expression more robustly in glial cells rather than in endothelial cells following TBI in a WT mice background, which we used in this study. It will be important to determine whether ApoA-I deficiency and the presence of Aâ together exacerbates TBI-related pathologies, especially given the interest in understanding TBI in the vulnerable elderly population (Thompson et al., 2006; Haring et al., 2015).
This study has several limitations. We only examined very acute time points of 6hr and 2D.Future studies are encouraged to include post-acute and chronic time points up to 6 months after injury, when potential secondary damage to endothelial cells (Villalba et al., 2017; Andrews, Lutton, Merkel, Razmpour, & Ramirez, 2016; Prakash & Carmichael, 2015) may enable effects of ApoA-I deficiency to be more easily observed. Further, more time points are needed to investigate the extent of modulated cytokine response that we have observed in 2D TBI ApoA-I KO animals, as it is unclear whether this response will persist beyond the 2D timeframe (Figure 2).
Despite these limitations, the study has several strengths, including the a priori blinding strategy used throughout. Based on the inflammatory changes presented thus far, future studies are encouraged to explore the mechanistic relationship between ApoA-I and neuroinflammation further, for example by supplementing ApoA-I KO TBI mice with HDL. To further characterize inflammatory outcomes observed in ApoA-I KO TBI mice, follow up work can be done to uncover transcriptomic changes induced by ApoA-I KO in the brain by techniques such as Single-Cell RNA-Seq (scRNAseq), which would help identify widespread changes in the brain beyond the capabilities of standard histology . Although this work is preliminary, our study shows that ApoA-I deficicency enhances acute inflammatory responses post a single CHIMERA TBI.The results from this study raises the possibility that ApoA-I supplementation may be of therapeutic value in TBI patients.,and shows how additional research in this area would be beneficial in advancing TBI treatment.
Acknowledgements
The authors would like to thank the Weston Brain Institute “for their support in funding for this project”.
References
- Andrews, A. M., Lutton, E. M., Merkel, S. F., Razmpour, R., & Ramirez, S. H. (2016). Mechanical Injury Induces Brain Endothelial-Derived Microvesicle Release: Implications for Cerebral Vascular Injury during Traumatic Brain Injury. Frontiers in Cellular Neuroscience, 10. https://doi.org/10.3389/fncel.2016.00043
- Balabanov, R., Goldman, H., Murphy, S., Pellizon, G., Owen, C., Rafols, J., & Dore-Duffy, P. (2001). Endothelial cell activation following moderate traumatic brain injury. Neurological Research, 23(2–3), 175–182. https://doi.org/10.1179/016164101101198514
- Bashir, A., Abebe, Z. A., McInnes, K. A., Button, E. B., Tatarnikov, I., Cheng, W. H., … Wellington, C. L. (2020). Increased severity of the CHIMERA model induces acute vascular injury, sub-acute deficits in memory recall, and chronic white matter gliosis. Experimental Neurology, 324, 113116. https://doi.org/10.1016/j.expneurol.2019.113116
- Besler, C., Lüscher, T. F., & Landmesser, U. (2012). Molecular mechanisms of vascular effects of High-density lipoprotein: Alterations in cardiovascular disease. EMBO Molecular Medicine, 4(4), 251–268. https://doi.org/10.1002/emmm.201200224
- Boyce, G., Button, E., Soo, S., & Wellington, C. (2017). The pleiotropic vasoprotective functions of high-density lipoproteins (HDL). Journal of Biomedical Research. https://doi.org/10.7555/JBR.31.20160103
- Button, E. B., Boyce, G. K., Wilkinson, A., Stukas, S., Hayat, A., Fan, J., … Wellington, C. L. (2019). ApoA-I deficiency increases cortical amyloid deposition, cerebral amyloid angiopathy, cortical and hippocampal astrogliosis, and amyloid-associated astrocyte reactivity in APP/PS1 mice. Alzheimer’s Research & Therapy, 11(1), 44. https://doi.org/10.1186/s13195-019-0497-9
- Calabresi, L., Franceschini, G., Sirtori, C. R., De Palma, A., Saresella, M., Ferrante, P., & Taramelli, D. (1997). Inhibition of VCAM-1 expression in endothelial cells by reconstituted high density lipoproteins. Biochemical and Biophysical Research Communications, 238(1), 61–65. https://doi.org/10.1006/bbrc.1997.7236
- Cheng, W. H., Martens, K. M., Bashir, A., Cheung, H., Stukas, S., Gibbs, E., … Wellington, C. L. (2019). CHIMERA repetitive mild traumatic brain injury induces chronic behavioural and neuropathological phenotypes in wild-type and APP/PS1 mice. Alzheimer’s Research & Therapy, 11(1), 6. https://doi.org/10.1186/s13195-018-0461-0
- Dietrich, J.-B. (2002). The adhesion molecule ICAM-1 and its regulation in relation with the blood-brain barrier. Journal of Neuroimmunology, 128(1–2), 58–68. https://doi.org/10.1016/s0165-5728(02)00114-5
- Elder, G. A., Gama Sosa, M. A., De Gasperi, R., Stone, J. R., Dickstein, D. L., Haghighi, F., … Ahlers, S. T. (2015). Vascular and Inflammatory Factors in the Pathophysiology of Blast-Induced Brain Injury. Frontiers in Neurology, 6. https://doi.org/10.3389/fneur.2015.00048
- Elliott, D. A., Weickert, C. S., & Garner, B. (2010). Apolipoproteins in the brain: Implications for neurological and psychiatric disorders. Clinical Lipidology, 51(4), 555–573. https://doi.org/10.2217/CLP.10.37
- George, N., & Geller, H. M. (2018). Extracellular matrix and traumatic brain injury. Journal of Neuroscience Research, 96(4), 573–588. https://doi.org/10.1002/jnr.24151
- Haring, R. S., Narang, K., Canner, J. K., Asemota, A. O., George, B. P., Selvarajah, S., Haider, A. H., & Schneider, E. B. (2015). Traumatic brain injury in the elderly: Morbidity and mortality trends and risk factors. Journal of Surgical Research, 195(1), 1–9. https://doi.org/10.1016/j.jss.2015.01.017
- Iqbal, A. J., Barrett, T. J., Taylor, L., McNeill, E., Manmadhan, A., Recio, C., … Fisher, E. A. (2016). Acute exposure to apolipoprotein A1 inhibits macrophage chemotaxis in vitro and monocyte recruitment in vivo. ELife, 5. https://doi.org/10.7554/eLife.15190
- Kenney, K., Amyot, F., Haber, M., Pronger, A., Bogoslovsky, T., Moore, C., & Diaz-Arrastia, R. (2016). Cerebral Vascular Injury in Traumatic Brain Injury. Experimental Neurology, 275 Pt 3, 353–366. https://doi.org/10.1016/j.expneurol.2015.05.019
- Lutton, E. M., Razmpour, R., Andrews, A. M., Cannella, L. A., Son, Y.-J., Shuvaev, V. V., … Ramirez, S. H. (2017). Acute administration of catalase targeted to ICAM-1 attenuates neuropathology in experimental traumatic brain injury. Scientific Reports, 7(1), 3846. https://doi.org/10.1038/s41598-017-03309-4
- Mahdy Ali, K., Wonnerth, A., Huber, K., & Wojta, J. (2012). Cardiovascular disease risk reduction by raising HDL cholesterol—Current therapies and future opportunities. British Journal of Pharmacology, 167(6), 1177–1194. https://doi.org/10.1111/j.1476-5381.2012.02081.x
- McKeating, E. G., & Andrews, P. J. (1998). Cytokines and adhesion molecules in acute brain injury. British Journal of Anaesthesia, 80(1), 77–84. https://doi.org/10.1093/bja/80.1.77
- Muoio, V., Persson, P. B., & Sendeski, M. M. (2014). The neurovascular unit – concept review. Acta Physiologica, 210(4), 790–798. https://doi.org/10.1111/apha.12250
- Namjoshi, D. R., Cheng, W. H., McInnes, K. A., Martens, K. M., Carr, M., Wilkinson, A., … Wellington, C. L. (2014). Merging pathology with biomechanics using CHIMERA (Closed-Head Impact Model of Engineered Rotational Acceleration): A novel, surgery-free model of traumatic brain injury. Molecular Neurodegeneration, 9, 55. https://doi.org/10.1186/1750-1326-9-55
- Prakash, R., & Carmichael, S. T. (2015). Blood-brain barrier breakdown and neovascularization processes after stroke and traumatic brain injury. Current Opinion in Neurology, 28(6), 556–564. https://doi.org/10.1097/WCO.0000000000000248
- Salehi, A., Zhang, J. H., & Obenaus, A. (2017). Response of the cerebral vasculature following traumatic brain injury. Journal of Cerebral Blood Flow and Metabolism: Official Journal of the International Society of Cerebral Blood Flow and Metabolism, 37(7), 2320–2339. https://doi.org/10.1177/0271678X17701460
- Schimmel, S. J., Acosta, S., & Lozano, D. (2017). Neuroinflammation in traumatic brain injury: A chronic response to an acute injury. Brain Circulation, 3(3), 135–142. https://doi.org/10.4103/bc.bc_18_17
- Shrikant, P., Weber, E., Jilling, T., & Benveniste, E. N. (1995). Intercellular adhesion molecule-1 gene expression by glial cells. Differential mechanisms of inhibition by IL-10 and IL-6. Journal of Immunology (Baltimore, Md.: 1950), 155(3), 1489–1501.
- Thompson, H. J., McCormick, W. C., & Kagan, S. H. (2006). Traumatic Brain Injury in Older Adults: Epidemiology, Outcomes, and Future Implications. Journal of the American Geriatrics Society, 54(10), 1590–1595. https://doi.org/10.1111/j.1532-5415.2006.00894.x
- Verbeek, M. M., Otte-Höller, I., Westphal, J. R., Wesseling, P., Ruiter, D. J., & de Waal, R. M. (1994). Accumulation of intercellular adhesion molecule-1 in senile plaques in brain tissue of patients with Alzheimer’s disease. The American Journal of Pathology, 144(1), 104–116.
- Villalba, N., Sackheim, A. M., Nunez, I. A., Hill-Eubanks, D. C., Nelson, M. T., Wellman, G. C., & Freeman, K. (2017). Traumatic Brain Injury Causes Endothelial Dysfunction in the Systemic Microcirculation through Arginase-1–Dependent Uncoupling of Endothelial Nitric Oxide Synthase. Journal of Neurotrauma, 34(1), 192–203. https://doi.org/10.1089/neu.2015.4340
- Vonder Haar, C., Martens, K. M., Bashir, A., McInnes, K. A., Cheng, W. H., Cheung, H., … Wellington, C. L. (2019). Repetitive closed-head impact model of engineered rotational acceleration (CHIMERA) injury in rats increases impulsivity, decreases dopaminergic innervation in the olfactory tubercle and generates white matter inflammation, tau phosphorylation and degeneration. Experimental Neurology, 317, 87–99.
- https://doi.org/10.1016/j.expneurol.2019.02.012 Yin, K., Deng, X., Mo, Z.-C., Zhao, G.-J., Jiang, J., Cui, L.-B., … Tang, C.-K. (2011). Tristetraprolin-dependent post-transcriptional regulation of inflammatory cytokine mRNA expression by apolipoprotein A-I: Role of ATP-binding membrane cassette transporter A1 and signal transducer and activator of transcription 3. The Journal of Biological Chemistry, 286(16), 13834–13845. https://doi.org/10.1074/jbc.M110.202275